
|
Dr. Prithviraj Bose
Case 1: Treatment of Anemia in Myelofibrosis
 |
Presenting Findings |
|
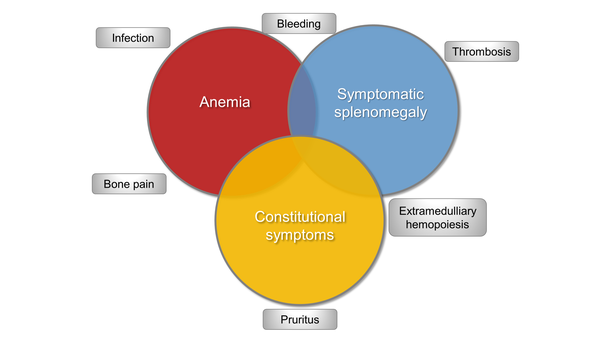
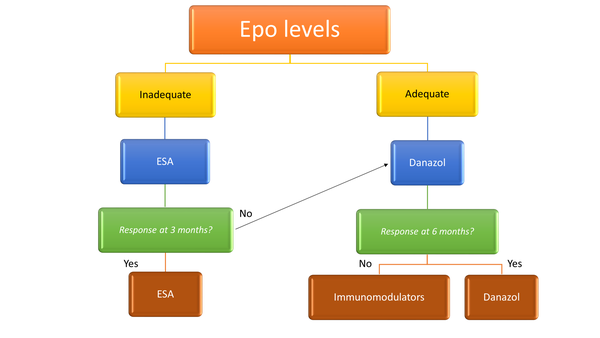
Anemia is one of the most common clinical manifestations of myelofibrosis (MF) (Figure 1).1 Erythropoietin (EPO) levels indicate whether or not the patient is likely to benefit from an erythropoietic stimulating agent (ESA) (Figure 2).1 Many patients will not benefit from the use of an ESA since they require too many transfusions or have too high of an endogenous EPO level, as the patient described in the above scenario. For this patient, the androgen danazol is appropriate, and before initiating this therapy in male patients, it is necessary to check prostate antigen levels for prostate cancer screening and to monitor liver function tests at baseline and over time. Danazol can be given at a dose of 200 mg 2 or 3 times a day, for at least 6 months to determine efficacy.
ESA=erythropoietin stimulating agent
Immunomodulatory drugs (IMiDs) such as lenalidomide, thalidomide, and pomalidomide are also effective in ameliorating anemia in MF. Another option for this patient is thalidomide at a low dose of 50 mg a day.1 Pomalidomide and lenalidomide can be combined with ruxolitinib, but although these agents can improve anemia, the combination can be rather myelosuppressive. Our group has investigated lenalidomide plus ruxolitinib in patients with MF-related anemia, and we found that this combination was difficult because of the myelosuppression.2 A German group is evaluating pomalidomide plus ruxolitinib in an ongoing trial for MF (NCT01644110).3,4
Myelofibrosis-related anemia remains an area of significant need and is the target of several drug development efforts. The bromodomain and extraterminal domain protein (BET) inhibitor CPI-0610 is a potentially disease-modifying agent that is being studied in the phase 2 MANIFEST study (NCT02158858) in treatment-naïve MF, and has been shown to improve anemia in this setting.5,6 Agents that are specifically under investigation for MF-related anemia include luspatercept, which is approved for treatment of anemia in patients with beta-thalassemia7 or myelodysplastic syndrome8; approvals were based on the proportion of patients achieving transfusion independence.7,8 This agent is a first-in-class erythroid maturation agent that binds several TGF-β superfamily ligands to diminish Smad2/3 signaling and enhance late-stage erythropoiesis.9 Luspatercept is being investigated for MF-related anemia in a phase 2 trial (Figure 3; NCT03194542).10,11 The primary endpoint for the transfusion-dependent patients was achieving transfusion independence, and those who were not transfusion dependent had to have an increase of hemoglobin of >1.5 g/dL over at least 12 weeks.
Figure 3. Study design of the phase 2 trial evaluating luspatercept for myelofibrosis-related anemia.10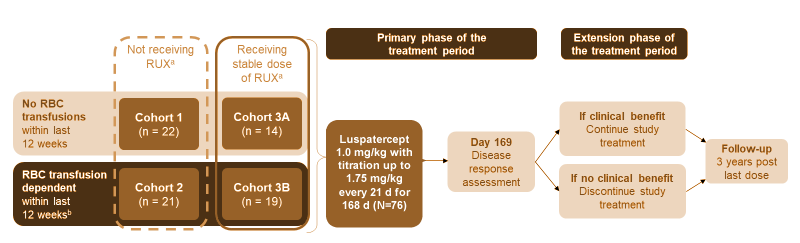
- No RBC transfusions (Cohorts 1 & 3A)
- – No RBC transfusions within 12 weeks before enrollment
- – Hb levels ≤ 9.5 g/dL (recorded on 3 separate occasions, with ≥ 14 d between each measurement)
- RBC transfusions (Cohorts 2 & 3B)
- – RBC transfusion dependent (2–4 RBC U/28 d within 12 weeks before enrollment)
- – No transfusion-free interval > 6 weeks
d=day; RUX=ruxolitinib; U=unit.
Results showed that luspatercept showed benefit in all the four cohorts, but especially in the cohort in which patients were on a stable dose of ruxolitinib and were also transfusion dependent (Figure 4).10 A phase 3 trial, INDEPENDENCE, is under development and focuses on this cohort. Editor’s note: At the time of this article’s publication, this trial was not yet open for enrollment. Please check www.ClinicalTrials.gov for updates.
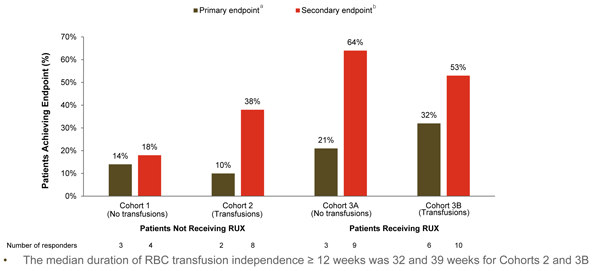
a For patients not receiving RBC transfusions: Hb increase ≥1.5 g/L at every assessment from baseline for ≥12 consecutive weeks, within the first 24 weeks on study. For patients receiving RBC transfusions: RBC transfusion free for ≥12 consecutive weeks, within the first 24 weeks on study.
b For patients not receiving RBC transfusions: mean Hb increase ≥1.5 g/dL from baseline for ≥12 consecutive weeks. For patients receiving RBC transfusions: ≥50% reduction in RBC transfusion burden from baseline.
RBC=red blood cell; Rux=ruxolitinib.
Similarly, sotatercept is a first-in-class, activin receptor IIA ligand trap that improves anemia by sequestering TGF-β superfamily ligands such as growth and differentiation factor 11 (GDF11), which suppress terminal erythroid differentiation.11 This agent is under phase 2 investigation both alone and in combination with ruxolitinib for treatment of MF-related anemia (NCT01712308).11,12 Both sotatercept and luspatercept are administered subcutaneously every 3 weeks and have been found to be very safe.10,11 Both agents have been associated with grade 3/4 adverse events such as hypertension and pain in the limb, bone, joint, or muscle,10,11 and diarrhea has also been reported with luspatercept,10 but these events occurred fairly infrequently.
Momelotinib is being evaluated vs danazol in the phase 3 MOMENTUM trial (Figure 5; NCT04173494).13 This JAK1/2 inhibitor has been observed to also inhibit ACVR1/ALK2, thus reducing production of hepcidin and increasing hemoglobin.
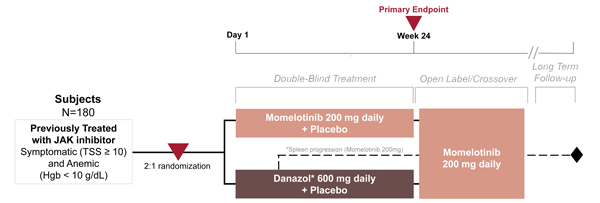
Global study: |
Danazol has been selected as an appropriate treatment comparator given its use to ameliorate anemia in myelofibrosis patients, as recommended by NCCN and ESMO guidelines. |
ESMO=European Society for Medical Oncology; Hgb=hemoglobin; NCCN=National Comprehensive Cancer Network; TSS=total symptom score.
Another ACVR1/ALK2 inhibitor, INCB000928, is under phase 1/2 testing in combination with ruxolitinib or as monotherapy for treatment of anemia due to myeloproliferative disorders (NCT04455841).14
Ruben A. Mesa, MD
Case 2: Treatment Considerations for Newly Diagnosed High-risk Polycythemia Vera
 |
Presenting Findings |
|
Considerations for treatment decision making are presented in Figure 6. When considering her risk of having polycythemia vera (PV), we note that this patient does not have a prior history of thrombotic or hemorrhagic event, but she is aged older than 60 years, and leukocytosis is present. She also does experience symptoms of disease, as assessed by the MPN 10. Based on her current laboratory results, we identify that the goals of therapy are to achieve phlebotomy independence over time, a white blood cell count lower than 10, a platelet count of less than 40,000, and to gain adequate control of symptoms. Another aspect of her treatment plan is to monitor for any signs of progression toward either MF (ie, enlarged spleen, changing symptoms, or fibrosis in the marrow) or more rarely, acute myeloid leukemia (AML). We determine that the patient has high-risk PV, based on her age and leukocytosis, and in terms of disease burden, she clearly is symptomatic. Based on the current National Comprehensive Cancer Network (NCCN) guidelines,15 the patient begins therapy with phlebotomies and aspirin, as well as cytoreductive therapy with hydroxyurea.
Figure 6. Considerations for treatment decision-making for patients with polycythemia vera.
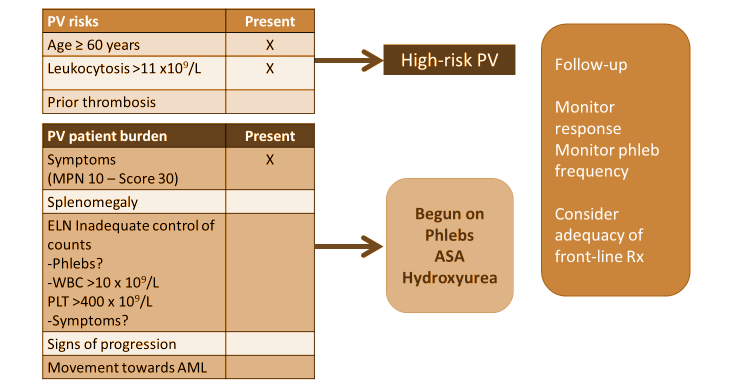
AML=acute myeloid leukemia; ASA=aspirin; phlebs=phlebotomies; PLT=platelet; PV=polycythemia vera; WBC=white blood cell count.
This is a standard case that illustrates some of the issues encountered in practice. Regarding risk assessment, patients look at the issue of age as a risk with some degree of skepticism. This patient is aged 61 years, just slightly higher than the 60 years numerical cutoff for a risk score.15 Patients may ask, “If I’m fit and 60, is that the same as not being fit and 60? Do I need to be on cytoreductive therapy?” However, the risk of having a vascular event in PV is real, and the ability to diminish that risk clearly has a significant beneficial impact, both in terms of decreasing that risk of thrombosis and bleeding, and also likely helps to improve the individual’s symptomatic burden that they face with the disease. In my practice, I discuss with patients that the goal for them is to have the disease be as invisible in their lives as much as possible—“invisible” meaning having good control of symptoms, being protected against the risk of vascular events, having no evidence of disease progression, and ideally tolerating therapy well so that it is not an ongoing detriment to their quality of life.
Baby aspirin at 81 mg daily (or 100 mg a day in Europe) has been shown in randomized trials to benefit patients with higher-risk PV to decrease the risk of vascular events almost irrespective of the other therapies the patient may be taking, as long as the patient is not allergic to aspirin.16 Achieving good control of hematocrit levels is an important factor in the treatment of this patient. A hematocrit level of 45% should be considered the ceiling for this value and not as a median, and thus, the goal is to maintain hematocrit levels below 45.17 In a trial led by Marchioli and coworkers,17 patients with PV were randomly assigned to receive intensive therapy (target hematocrit, >45%; low-hematocrit group) or less intensive therapy (target hematocrit: 45%-50%; high-hematocrit group). The primary endpoint was time until death from cardiovascular causes or major thromboembolic events; the secondary endpoints included total cardiovascular events. Results are presented in Figure 7 and support the need to target hematocrit of >45% when managing patients with PV.
Figure 7. Kaplan-Meier curves for the primary endpoint and total cardiovascular events in patients with polycythemia vera receiving either intensive therapy or less intensive therapy.17
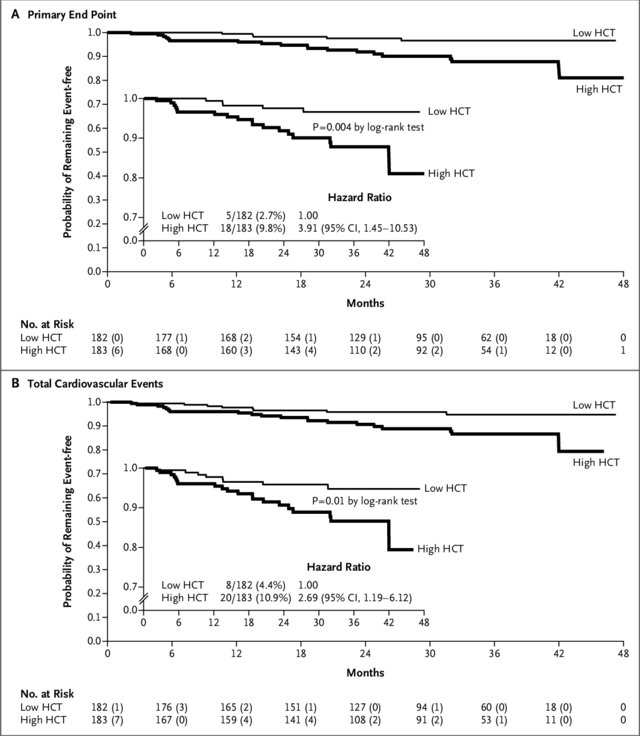
A) Time to the primary endpoint, eg, death from cardiovascular causes or thromboembolic events; and B) total cardiovascular events, eg, primary endpoint plus superficial-vein thrombosis (secondary endpoint), between the low- and high-hematocrit cohorts. The inset graphs present a detailed version of the overall graphs down to a probability of 0.7. Hazard ratios were calculated using a Cox proportional-hazards model.
CI=confidence interval; HCT=hematocrit.
The last factor of treatment for this patient is cytoreductive therapy, which is beneficial for a majority of patients. For frontline cytoreductive therapy, the NCCN recommends hydroxyurea, interferon alfa, or peginterferon alfa.15 Patients who should not be on cytoreductive therapy include those aged younger than 60 years and those with no significant leukocytosis, no significant symptom burden on phlebotomy and aspirin, and no history of vascular events who are tolerating phlebotomy well. This minority of patients will most likely do well on phlebotomy and aspirin alone.
In historical trials, hydroxyurea was shown to decrease vascular events vs phlebotomy alone,18 but has also been associated with toxicities such as cytopenias, mouth ulcers, leg ulcers, and an increased risk for lung cancer.19,20 There is also much discussion whether hydroxyurea increases the long-term risk of AML. Patients who are not candidates for cytoreductive therapy with hydroxyurea include younger individuals, particularly if they might become pregnant while on the therapy, and fever has been observed in women of childbearing potential. Patients with a history of skin ulcers or skin cancers, and those with neutropenia may also not be suitable candidates for hydroxyurea therapy. Finally, patients with more aggressive disease with splenomegaly and other features may not achieve adequate control with hydroxyurea. However, for many patients, hydroxyurea is the treatment choice, as many patients do well in this therapy, and it is an inexpensive drug.
The NCCN guidelines also recommends interferon alfa or peginterferon alfa.15 Ropegylated interferon may be another option, as a Biologic License Application has been submitted to the FDA in June 2020.21 Interferons have been shown to help to control the counts in randomized studies22-24 for at least one year, and may be superior over a longer period of time in decreasing the allele burden and potentially improving the risk of disease progression.25,26 Interferon therapy is preferred in younger individuals, especially in women of childbearing potential who might become pregnant.15 Toxicities can include flu-like symptoms, malaise, injection site reactions, autoimmune manifestations, and depression.
Once the patient begins therapy, we monitor for toxicity and response, and for our patient, we would monitor her over time to determine if the need for phlebotomy decreases significantly, if her hematocrit levels stay below 45%, if the white count clearly falls below 10, if the platelets are controlled, and if the symptoms improve or resolve. We also will monitor the patient to ensure that the disease is becoming invisible in her life as much as possible, and if she is tolerating the therapy well. In my opinion, one unmet goal is that patients may be treated with their maximum tolerated dose of hydroxyurea, but not a dose that has been efficacious for them. Thus, if they are taking hydroxyurea at a dose of 500 mg a day or 500 mg twice a day, but still the white count and platelets are high, and if they are still having symptoms, that is not an optimal response. In those situations, we need to consider a second-line therapy potentially with ruxolitinib, which is approved in the second-line setting, or depending upon the scenario if it's a purely cytoreductive inadequate benefits, you might still consider moving to the interferon as alternative.15
There some ongoing clinical trials looking at frontline treatment of PV. PTG-300 is a hepcidin agonist that is under evaluation in pre-high-risk patients as an alternative to phlebotomy (NCT04057040).27 Other frontline studies include those investigating interferon in patients with lower-risk PV, such as the low-risk PV study that was done in Italy and was presented earlier this summer at the European Hematology Association meeting.28,29 Findings suggest that interferon was superior to phlebotomy and aspirin in patients with lower-risk PV. For individuals that are failing frontline therapies, MDM-2 inhibitors are being studied as potentially being active in this cohort of patients (NCT03662126).30,31
In conclusion, when treating the patient with PV, we should consider their risks, as well as their disease burden when designing a treatment plan. We should discuss with the patient and layer that treatment plan in terms of that baseline of phlebotomy and aspirin and hematocrit control. We should aim to achieve true hematocrit control and consider appropriate use of cytoreductive therapy, likely in the majority patients with PV, and very importantly, monitor those patients for adequate response. We now have good options in the second-line setting, and the appropriate identification of patients that might benefit from a change in therapy is a key appropriate part of managing patients with PV. I do think that we will have multiple new agents that will be in our arsenal to help to treat patients with PV in the future, and I'm very hopeful for the future for these patients.